Sabemos que mesmo em um período tão especial, muitas dúvidas podem surgir, não é mesmo?
Então, que tal algumas dicas para as mamães de primeira viagem? Anote aí:
Cuide da sua saúde física e mental;
Faça uma lista com tudo que você precisa comprar para evitar aquele esquecimento e sufoco de última hora;
Busque informações sobre amamentação;
Faça o seu plano de parto e, se for necessário, não hesite em tirar todas a dúvidas sobre o momento da chegada do bebê com o obstetra;
Se precisar, peça ajuda! Conte com quem você confia, seu parceiro, familiares, médicos e todas as pessoas que você se sente confortável para ajudar, caso precise;
Visite obstetras, maternidades e pediatras com antecedência para identificar aqueles que você mais confia.
Gostou das dicas? Salve as dicas para conferir sempre que precisar!
A maioria das crianças cardiopatas (86%) nascem de mães consideradas de baixo risco, de pré-natal habitual. Ainda são poucos os diagnósticos fetais das cardiopatias. A maior parte desses bebês nasce sem o diagnóstico e um tempo precioso é desperdiçado muitas vezes até a descoberta da alteração, interferindo diretamente na qualidade do atendimento.
O ultrassom morfológico realiza um “screening” do coração, ou seja, funciona como um rastreio. O ecocardiograma fetal é um estudo mais aprofundado, onde avaliamos toda a anatomia e função cardíaca de maneira detalhada e precisa, e que chega a ter acurácia de até 98% no diagnóstico das alterações. O ecocardiografista fetal trabalha em parceria com toda a equipe de obstetrícia e ultrassonografia.
É um exame realizado através de equipamento de ultrassom, especialmente ajustado para a análise do coração fetal. Toda a estrutura anatômica, forma, função e ritmo cardíacos, serão avaliados de forma extremamente detalhada, para que nenhum diagnóstico importante fique sem ser conhecido. Realizado por profissional especialista em coração fetal, é ferramenta precisa e necessária para o reconhecimento das várias formas de cardiopatias, que, apesar de serem as alterações mais prevalentes no feto, não são as mais diagnosticadas por ultrassom convencional (morfológico ou obstétrico).
A nossa missão é oferecer uma assistência de excelência em Medicina Materno Fetal e Ultrassonografia em Ginecologia e Obstetrícia , que alia acolhimento, respeito, ética, conhecimento embasado nas melhores evidências científicas, uma equipe qualificada e tecnologia para rastrear, diagnosticar, prevenir e tratar doenças que podem afetar a gestação.
Nossa unidade Santo Agostinho foi desenhada para te fazer sentir em casa em uma etapa tão importante da sua vida!
Para agendamentos entre em contato com nossa equipe!
A Medicina Fetal é especialidade que tem o desafio de cuidar da vida do homem em um cenário que não é possível contato direto – o feto em desenvolvimento no útero. É desafiador e ao mesmo tempo admirável a possibilidade de intervir em uma fase muito precoce da vida com o avanço das técnicas e tratamentos desenvolvidos nos últimos 25 anos. Tais intervenções podem transformar radicalmente a vida daquele ser que está por nascer. Exemplos disso são as cirurgias fetais, como a reparação da mielomeningocele ainda no útero materno. Mas apesar desse êxito da medicina, existem algumas malformações e doenças fetais que não tem possibilidade de tratamento, nem após o nascimento. O comunicado do diagnóstico aos pais de que aquele bebe tem uma alteração é uma etapa da assistência médica muito delicada.
Como a maioria dos médicos, meu treinamento na faculdade e residência médica foi dedicado ao diagnóstico de doenças e ao domínio de opções terapêuticas. Não estamos preparados para ficar sem escolhas terapêuticas. E quando nos encontramos diante de um diagnóstico que não existe tratamento ou que apesar dos nossos esforços não somos capazes de chegar a um resultado satisfatório, experimentamos, em um primeiro momento, a sensação de fracasso, de impotência, de escassez e isso pode causar ruídos de comunicação entre médico e paciente.
O momento de falar sobre um diagnóstico que vai contra as expectativas dos pais de um filho perfeito e saudável é delicado, pois naquele instante estamos desconstruindo toda uma ilusão criada em torno daquela gestação e daquele bebe. Naquele instante… desconstruímos um sonho. O diagnóstico de uma síndrome genética ou malformação fetal que não permitem a sobrevida daquela criança após o nascimento ou que permita uma vida que não vai de encontro as expectativas dos pais, é catastrófico. Qual seria a melhor maneira do médico comunicar o diagnóstico e amenizar esse sofrimento dos pais? É uma pergunta muito difícil… Não existe uma regra. Cada família terá sua demanda e cada patologia tem suas peculiaridades. Mas é certo que quando nos colocamos de lado da paciente e da família, ela sente que nesse momento compartilhamos da sua dor, sofremos um pouco o sofrimento dela, percebe que sua tristeza foi vista e reconhecida, que a existência de seu filho, ainda que breve, está sendo respeitada e admirada. Não ter opções terapêuticas não é sinônimo de não se ter mais nada a oferecer. O efeito de apenas estar ali compartilhando e sendo solidário é incrível, é curativo para ambos os lados! Nesse momento somos ponte entre dois mundos: o da expectativa de uma criança perfeita e o da nova realidade que se apresenta. Devemos ser essa ponte estando presentes de fato, compartilhando com o casal de forma respeitosa e carinhosa as evidências cientificas existentes até então e acolhendo com empatia e escuta.
Médicos e pacientes precisam aprender a reconhecer os limites do poder da medicina. E isso significa ter compaixão por nós mesmos. Lutamos diariamente para conciliar a grande responsabilidade de curar com a realidade de que toda vida é finita. A beleza da vida não deveria ser tomada como garantida, existe o outro lado… existe a escuridão. Mas essa escuridão nos permite ver a luz, quando compreendemos e convivemos com sabedoria e leveza com nossos limites.
A empatia, a comunicação e a tomada de decisão compartilhada são essenciais e tem cada vez mais valor na nossa sociedade. Eu acredito que esse é o caminho para uma relação de respeito e informação.
Sejamos essa ponte, pelas nossas pacientes e por nós!
Texto de autoria da Dra. Marianna Amaral Pedroso.
Baseado em uma profunda conversa com um grande amigo durante um voo e no relato de Bettina Paek “The Unexpected Power of Presence”, publicado no ACOG em abril de 2019
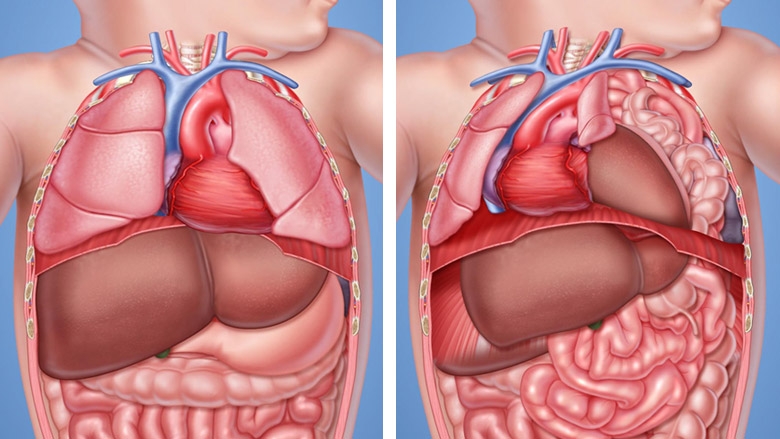
A hérnia diafragmática congênita (HDC) é uma condição rara, que afeta aproximadamente 1 a cada 4000 nascidos vivos. Essa doença ocorre por uma malformação no diafragma (musculatura que separa o nosso tórax do abdome). Ocorre uma falha no fechamento dessa musculatura durante o desenvolvimento do feto, permitindo a comunicação do tórax com o abdome. Com isso, dependendo do tamanho dessa comunicação, os orgãos abdominai, como por exemplo o estômago, intestino e fígado, podem passar por essa falha no diafragma e ocupar parte do tórax do bebê.
Atualmente, com o avanço da Medicina Fetal, dois terços dos casos são detectados ainda no pré-natal por meio da ultrassonografia já no primeiro trimestre e, especialmente, no 2º trimestre de gestação.
Essa anomalia congênita causa muitas dúvidas nas gestantes que recebem a notícia que o seu bebê tem uma HDC. Por conta disso, esse post visa esclarecer algumas dúvidas sobre esse tema.
Quais as consequências da Hérnia Diafragmática Congênita para o bebê?
A HDC está associada a alta mortalidade e morbidade da criança afetada. Com os órgãos abdominais ocupando parte do tórax, o pulmão do bebê não se desenvolve da maneira correta, acarretando no que chamamos de Hipoplasia Pulmonar. Com isso, o bebê ao nascer terá um pulmão pouco desenvolvido e, como consequência, terá dificuldade de oxigenação.
Além disso, com um pulmão pequeno a pressão do sangue nas artérias do pulmão fica aumentada, o que pode causar um quadro chamado de Hipertensão Pulmonar. Assim, a maioria dos bebês após o nascimento não consegue respirar sozinhos e precisa receber cuidados especializados em uma UTI neonatal.
Entretanto, todas essas consequências da HDC variam de caso a caso, pois tudo depende do volume ocupado por certos órgãos no tórax do bebê. Assim, logo após o diagnóstico é de extrema importância que a gestante tenha acesso a uma equipe de Medicina Fetal para ter uma avaliação especializada que determinará a gravidade da HDC e encaminhará a gestante para um centro especializado no tratamento dessa alteração fetal, caso seja indicado.
Como determinar a severidade da Hérnia Diafragmática Congênita?
Por meio de um ultrassom realizado por um especialista altamente capacitado (fetólogo) ou por uma ressonância magnética será avaliado:
Relação pulmão-cabeça (RPC): é uma relação entre a área do pulmão contralateral à hérnia e a circunferência da cabeça do bebê. Um valor abaixo de 1 é indicativo de pior prognóstico.
Posição do fígado: visa avaliar se o fígado está ou não ocupando a cavidade torácica, e caso afirmativo, a medida do volume ocupado. A presença do fígado no tórax está associada a um pior prognóstico.
Posição do estômago: será avaliado se o estômago está ocupando espaço na cavidade torácica e qual a posiçao dele no tórax. Dependendo dessa posição há pior ou melhor prognóstico da hérnia.
Presença de anomalias associadas: cerca de 1/3 dos bebês com HDC tem problemas cardíacos associados. Por esse motivo, todos esses bebês devem realizar um ecocardiograma fetal. Outras anomalias associadas são problemas no trato urinário, no sistema nervoso central, malformações gastrointestinais, dentre outras menos comuns. A presença de outras alterações estruturais e/ou genéticas associadas pioram o prognóstico.
Além disso, é aconselhado investigação genética dos fetos que tem a HDC. Essa avaliação do DNA do bebê busca alterações cromossômicas ou mutações gênicas que possam explicar o aparecimento da HDC no feto. Em 40% dos casos, a HDC acontece associada a outras alterações anatômicas que aumentam o risco de uma síndrome genética naquela criança.
HDC esquerda e HDC direita: qual é a diferença?
Aproximadamente 83% dos bebês com HDC têm um defeito no lado esquerdo do diafragma. Uma HDC do lado esquerdo permite que estômago, intestinos e, às vezes, o fígado se mova (hernie) para dentro do tórax do bebê.
Os outros 17% dos bebês com HDC apresentam um defeito no lado direito do diafragma. Uma HDC do lado direito quase sempre permite que o fígado se mova para o tórax.
Para garantir um diagnóstico preciso, é importante visitar um centro de terapia fetal com uma equipe multidisciplinar com experiência na avaliação de gestações afetadas pela HDC. Este é um passo importante para determinar as opções de tratamento disponíveis e garantir os melhores resultados possíveis para o bebê.
Manejo da Hérnia Diafragmática Congênita durante a gestação
A HDC tem tratamento, mas a sua correção só é realizada após o nascimento do bebê por um cirurgião pediátrico. Já a conduta durante o pré-natal vai depender muito da gravidade da hérnia.
Para hérnias em que o fígado não está ocupando a cavidade torácica e que o pulmão do bebê está se desenvolvendo de maneira adequada, a conduta durante o pré-natal é expectante. O médico especialista em Medicina Fetal realizará uma monitorizarão do bebê durante o pré-natal para acompanhar a evolução da HDC e a vitalidade do bebê e, após o nascimento, a criança será estabilizada em uma UTI neonatal e só então operada para correção da falha no diafragma.
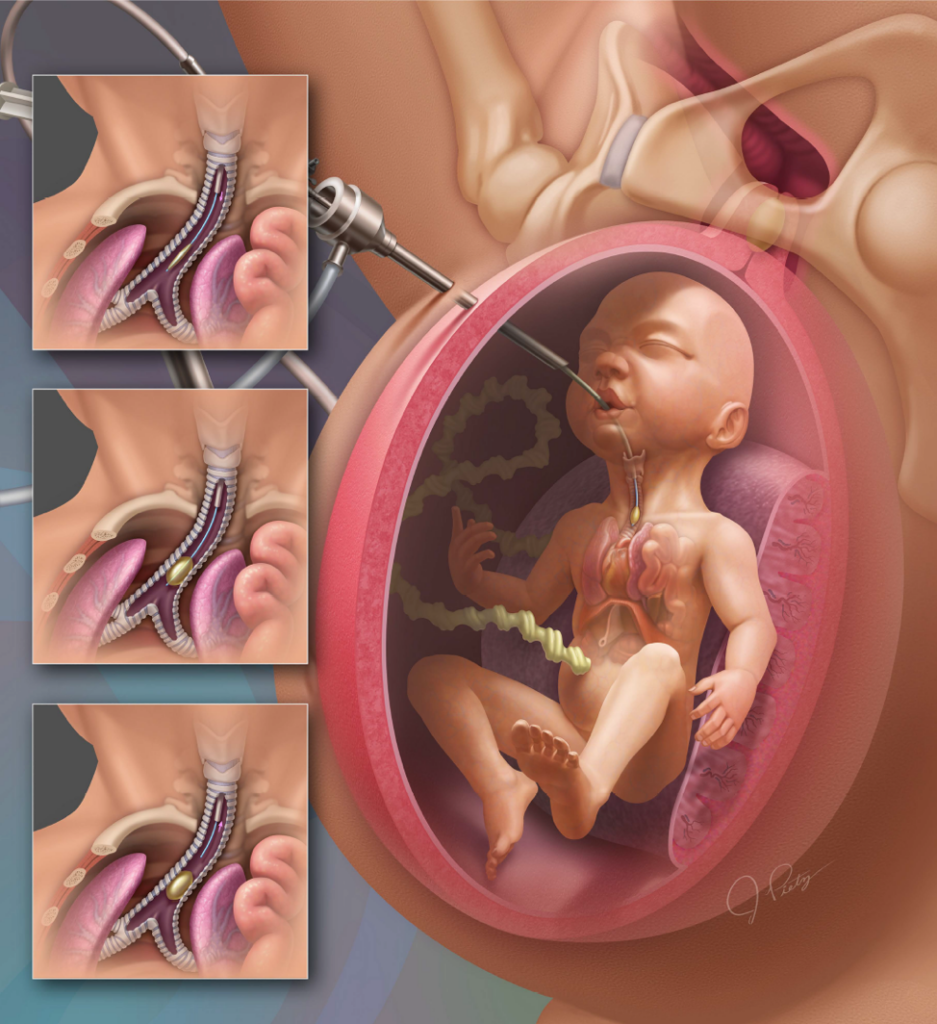
Já em casos graves, com herniação do fígado e uma relação pulmão/ cabeça menor que 1 existe a possibilidade de realizar uma cirurgia intraútero chamada de Oclusão Traqueal Fetal. Esse procedimento é minimamente invasivo, deve ser realizada em torno de 26 – 28 semanas de gestação e o bebê não pode ter outras alterações anatômicas ou genéticas que piorem o prognóstico pós-natal.
A Oclusão Traqueal Fetal consiste na colocação de um balão na traquéia do feto por meio de um aparelho chamado fetoscópio. Esse balão irá ocluir a traquéia impedindo a saída do líquido produzido pelos pulmões. Essa obstrução das vias aéreas intraútero irá estimular o desenvolvimento pulmonar. Antes que você se assuste, isso não fará mal ao feto, pois o bebê no útero recebe oxigênio pelo cordão umbilical! Em seguida, com cerca de 34 semanas (ou após 6 semanas da cirurgia) esse balão é retirado e posteriormente o bebê poderá nascer por meio de uma cesariana ou por parto normal.
“O objetivo dessa cirurgia é melhorar o desenvolvimento pulmonar do bebê e aumentar as chances de sobrevivência após o nascimento.”
Agora, com todo esse conhecimento, sabe-se que após uma gestante ter o diagnóstico de um bebê com HDC, ela deve buscar sempre um serviço altamente especializado nesse assunto. Assim, por meio de uma equipe multidisciplinar, composta por profissionais da medicina fetal, cirurgiões pediátricos e neotatologistas as chances de sobrevivência do bebê serão muito maiores.
Figura ilustrativa demonstrando a entubação intrautero do feto para oclusão traqueal com balão no tratamento da Hérnia Diafragmática Congênita.
Qualquer dúvida a respeito desse assunto entre em contato conosco que iremos ajudá-los da melhor maneira possível!
Texto de autoria da Dra. Marianna Pedroso.
Referências:
CORDIER, A.-G., RUSSO, F. M., DEPREST, J., & BENACHI, A. (2019). Prenatal diagnosis, imaging, and prognosis in Congenital Diaphragmatic Hernia. Seminars in Perinatology.doi:10.1053/j.semperi.2019.07.002
Deprest, J., Gratacos, E., & Nicolaides, K. H. (2004). Fetoscopic tracheal occlusion (FETO) for severe congenital diaphragmatic hernia: evolution of a technique and preliminary results. Ultrasound in Obstetrics and Gynecology,
Russo, F. M., Cordier, A.-G., De Catte, L., Saada, J., Benachi, A., & Deprest, J. (2018). Proposal for standardized prenatal ultrasound assessment of the fetus with congenital diaphragmatic hernia by the European reference network on rare inherited and congenital anomalies (ERNICA). Prenatal Diagnosis, 38(9),
A amniocentese é um procedimento médico em que é coletado uma pequena quantidade de líquido amniótico, que é o fluido que fica ao redor do bebê no útero, por meio de uma agulha. Essa amostra de líquido é posteriormente enviada a um laboratório para realizar testes diagnósticos, como por exemplo, avaliação da genética do feto para saber se tem Síndrome de Down ou outras anomalias genéticas.
Esse procedimento geralmente é realizado quando há alguma suspeita de alterações na anatomia do bebê pelo ultrassom de rotina ou pela detecção de marcadores ultrassonográficos que aumentam o risco da criança ter alguma síndrome genética. Também pode ser realizado caso haja uma doença hereditária na família e há o desejo de investigar se o feto foi afetado por essa doença, para investigação de infecção intraútero e até mesmo para teste de paternidade.
A seguir algumas dúvidas que muitas gestantes tem ao realizar esse exame:
Como a amniocentese é realizada?
A amniocentese é realizada por um médico especialista e sempre é feita guiada por um ultrassom. Não há necessidade de realizar nenhum preparo especial antes do exame, isso significa que você pode comer e beber normalmente no dia.
Primeiro o médico vai checar a posição do bebê e conferir os batimentos cardíacos fetais no ultrassom. Em seguida, vai limpar a sua pele do abdome com um sabão anti-séptico. Depois, uma agulha é inserida no seu abdome em direção ao útero a fim de alcançar o líquido amniótico e é retirada uma amostra de 15-30 ml. Todo o procedimento é feito guiado pelo ultrassom e é um procedimento que não machuca o bebê. Essa agulha fica dentro do seu útero só por alguns minutos.
Durante o procedimento não há a necessidade de nenhum tipo de anestésico, e você ficará acordada o tempo todo. Alguns médicos realizam um botão anestésico no local onde será inserido a agulha no abdome para mais conforto da gestante. No final o médico vai colocar um curativo no local que foi realizada a punção. Se você se sentir insegura, pode trazer um acompanhante para te dar suporte emocional durante o procedimento.
A amniocentese dói?
A dor da amniocentese é similar à de tirar sangue, mas você também pode sentir cólicas durante o exame. A maioria das mulheres não precisa de nenhuma medicação para realizar o procedimento.
Quais são os riscos?
Toda gestação tem um pequeno risco de evoluir para aborto espontâneo e realizar uma amniocentese pode aumentar um pouco esse risco. Essa complicação acontece em 1 a cada 200 gestantes, ou seja, o risco é menor que 0,5%.
Além disso, é importante explicar que a quantidade de líquido amniótico retirada durante o exame é muito pequena e não afeta o desenvolvimento do bebê se realizada após as 16 semanas.
Preciso realizar algum exame antes de fazer a amniocentese?
Antes do procedimento o médico deve saber a sua idade gestacional. Como já foi falado, a amniocentese não deve ser feita antes de 16 semanas de gestação, devido aos maiores riscos de complicações. Além disso, é importante você saber qual é o seu grupo sanguíneo, pois se for negativo (Rh -), após a amniocentese você precisará tomar uma vacina anti-imunoglobulina D. Outros exames que devem ser realizados são testes para hepatite B e HIV. Se você tiver alguma outra doença você deve discutir com seu médico a necessidade de realizar outros exames.
Quanto tempo eu fico de repouso após o procedimento?
Após a amniocentese deve-se evitar a prática de atividade física por 1-2 dias, contudo não há necessidade de repouso absoluto. É comum sentir uma dor leve na primeira noite após o exame, você pode tomar um comprimido de analgésico recomendado pelo médico assistente, se sentir necessidade.
Quando eu devo buscar atendimento médico após a amniocentese?
Se você tiver algum sangramento vaginal, dor abdominal importante, febre alta ou perda de líquido na vagina você deve ir a um pronto atendimento. Em geral, se você permanecer bem após a 1 a 2 semanas após a amniocentese, é incomum que complicações aconteçam.
Como eu sei sobre os resultados da amniocentese?
O seu médico vai marcar uma consulta de retorno para que ele te explique os resultados. Além disso, você deve passar o seu telefone para que se alguma alteração importante for detectada você seja comunicada mais brevemente.
Dependendo do laboratório em que o material será analisado, o resultado será encaminhado diretamente para você e tão logo isso aconteça é importante agendar uma consulta com seu médico assistente para esclarecimento de dúvidas.
Essas são algumas dúvidas comuns a respeito da amniocentese. Lembre-se que explicamos de uma maneira geral, mas cada caso deve ser avaliado de maneira individualizada.
A trissomia do 18, designada pelo epônimo síndrome de Edwards, é uma síndrome genética cuja incidência de 1: 3000-7000 nascidos vivos, sendo três vezes mais frequente no sexo feminino. Corresponde à segunda trissomia autossômica mais comum na população recém-nascida, logo após a síndrome de Down (trissomia do 21). Existe correlação entre a idade materna avançada e a ocorrência da trissomia do 18. Ademais, a taxa de recorrência da doença (probabilidade uma mulher com feto prévio diagnosticado com síndrome de Edwards gestar outro filho com a doença) gira em torno de 1%.
Sabe-se que a maioria dos portadores da síndrome de Edwards apresenta uma cópia extra do cromossomo 18 inserida no genoma de todas as suas células. Esse material genético supranumerário é responsável por desencadear múltiplas alterações no desenvolvimento de diversos órgãos do corpo humano.
A suspeita de trissomia do 18, bem como de outras anormalidades cromossômicas, costuma advir de alterações morfológicas do feto detectadas ao ultrassom. O tipo e a frequência desses achados ultrassonográficos variam de acordo com a idade gestacional, sendo que os mais documentados são: restrição de crescimento intrauterino (feto pequeno para a idade gestacional), defeitos de fechamento da parede abdominal, cardiopatias, cistos do plexo coróide, aumento da translucência nucal, anormalidades cranianas variadas, posicionamento anormal das mãos (mãos cerradas), entre outros.
Atualmente, o rastreamento de síndromes genéticas no feto é realizado precocemente por meio da avaliação de marcadores ecográficos no exame do primeiro trimestre (entre 11 e 13 semanas e 6 dias). Esses marcadores são: translucência nucal, osso nasal ausente ou hipoplásico, regurgitação tricúspide e avaliação do ducto venoso. A avaliação de todos esses marcadores associado a avaliação da anatomia fetal e história materna tem uma taxa de detecção de aproximadamente 90% das síndromes genéticas, dentre elas a trissomia do 18.
O NIPT, exame que avalia fragmentos de DNA fetal no sangue materno, é outro método de rastreamento, com sensibilidade muito grande para síndromes genéticas, principalmente a trissomia do 21, 18 e 13.
Vale ressaltar que o diagnóstico é dado por meio da análise do cariótipo fetal. Obtém-se uma amostra de células fetais ou placentárias (por amniocentese, biópsia de vilo corial ou cordocentese) e sua análise permite contar e enumerar os pares de cromossomos.
Geralmente a doença cursa com prognóstico reservado e muitos estudos apontam a morte intraútero como um desfecho frequente durante o pré-natal. As doenças cardíacas congênitas ocorrem em mais de 50% dos portadores da síndrome e o acometimento gastrointestinal está presente em aproximadamente 75%. Em geral, 50% dos bebês nascidos com a síndrome morrem nas duas primeiras semanas de vida e em torno de 5-10% sobrevivem ao primeiro ano de idade. Os principais problemas médicos descritos para os sobreviventes de longa data incluem escoliose, refluxo gastroesofágico, perda auditiva e tumor de Wilms, além de défict intelectual significativo. Curiosamente, os estudos científicos apontam que os portadores do sexo feminino têm maior chance de sobreviver durante a gestação e após o nascimento, isso justifica a maior ocorrência da síndrome em pacientes femininas e uma expectativa média de vida mais longa estar associada a esse sexo (9,6 meses de vida são esperados para o sexo feminino e 1,4 meses para os pacientes masculinos).
Como dito anteriormente, o prognóstico da trissomia do 18 costuma ser desfavorável e nenhum tratamento atual mostrou-se capaz de curar ou prevenir a doença. Nesse sentido, o diagnóstico pré-natal é importante porque permite que os pais e o obstetra se planejem em relação aos cuidados com pré-natal, parto e o recém-nascido. O aborto de fetos que tem síndromes genéticas não é permitido no Brasil. Após o nascimento do bebê, a família, em sintonia com a equipe médica, pode optar por uma abordagem terapêutica mais paliativa ou intervencionista. As possíveis intervenções médicas a serem realizadas têm como objetivo corrigir defeitos graves e ameaçadores da vida, sua disponibilidade depende de quais anormalidades o feto apresenta. Em todos os casos, é imprescindível uma abordagem multidisciplinar e individualizada do paciente que tem a trissomia do 18!